
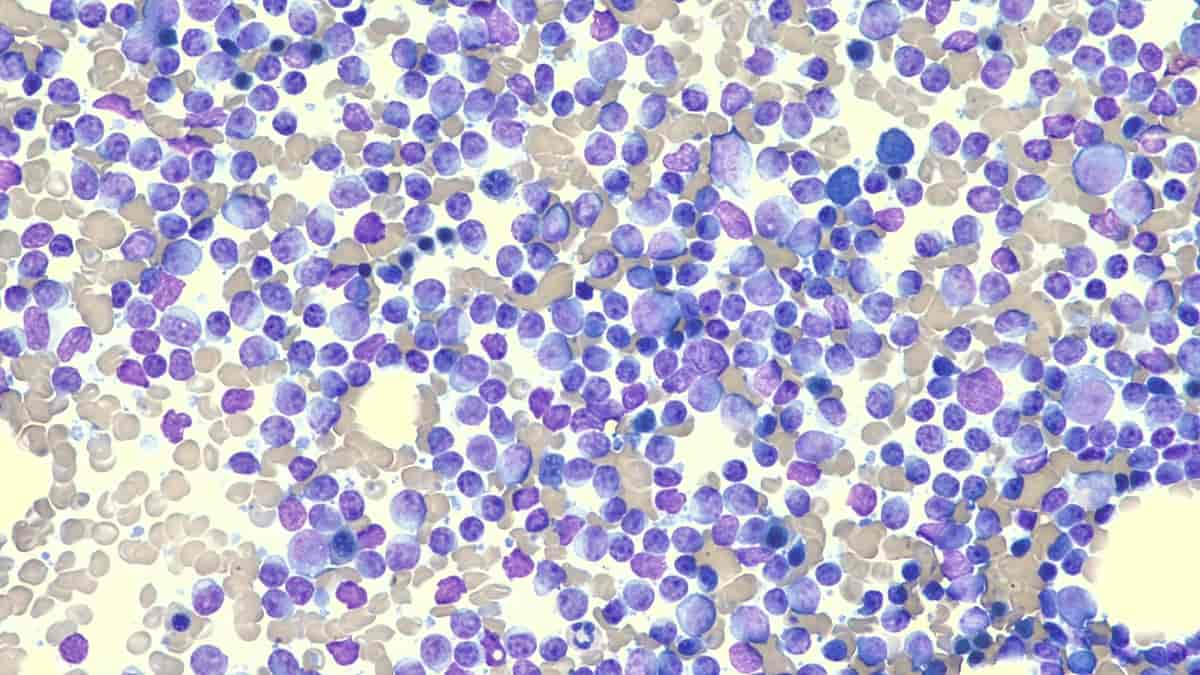
Kronisk lymfatisk leukemi (KLL) er en blodkreftsykdom som har sitt opphav i lymfocyttene, som er en undergruppe av de hvite blodcellene i beinmargen. Det er den vanligste typen blodkreft, og hvert år får 300–350 personer diagnosen i Norge.
Faktaboks
- Uttale
-
lymf'atisk leukem'i
- Også kjent som
-
KLL
engelsk: chronic lymphocytic leukaemia (CLL)
Årsaken er ikke kjent, men det ligger en svak disposisjon til å få sykdommen i genene (polygen arv). Diagnosen stilles med blodprøve. Hovedkriteriet for å få diagnosen kronisk lymfatisk leukemi (KLL) er at det er mer enn fem milliarder lymfocytter per liter (5 x 109/l) i blodet, og at disse har kjennetegnene som er typisk for kreftcellen. Hvis man påviser slike kreftceller i lymfeknute og det er mindre enn 5 x 109/l av dem i blod kalles sykdommen småcellet lymfocytært lymfom (SLL). SLL og KLL er ellers samme sykdom. Det er som regel ingen symptomer knyttet til KLL før sykdommen blir mer alvorlig. Da kan man bli slapp, få anemi, bli mer utsatt for infeksjoner og få forstørrede lymfeknuter.
Det finnes en rekke medikamenter mot KLL, for eksempel cellegift, antistoffer og enzym-hemmere.

Kommentarer
Kommentarer til artikkelen blir synlig for alle. Ikke skriv inn sensitive opplysninger, for eksempel helseopplysninger. Fagansvarlig eller redaktør svarer når de kan. Det kan ta tid før du får svar.
Du må være logget inn for å kommentere.