Pasienter med forstørret milt og høye verdier av hvite blodceller ble i 1840-årene beskrevet i Frankrike, Skottland og Tyskland. Disse hadde sannsynligvis KML. Den fulle forståelsen av den molekylære årsaken til sykdommen kom imidlertid etter en trinnvis utvikling som startet i 1960 da de to amerikanske kreftforskerne Peter Nowell (1928–2016) og David Hungerford (1927–1993) beskrev syv pasienter med KML-lignende sykdom som alle hadde et kromosom 22 med forkortelse av den ene armen. Dette gjaldt bare leukemicellene, mens andre celler hadde et normalt kromosom. Det lille kromosom 22 ble heretter hetende Philadelphia-kromosomet fordi oppdagelsen ble gjort i byen Philadelphia i USA.
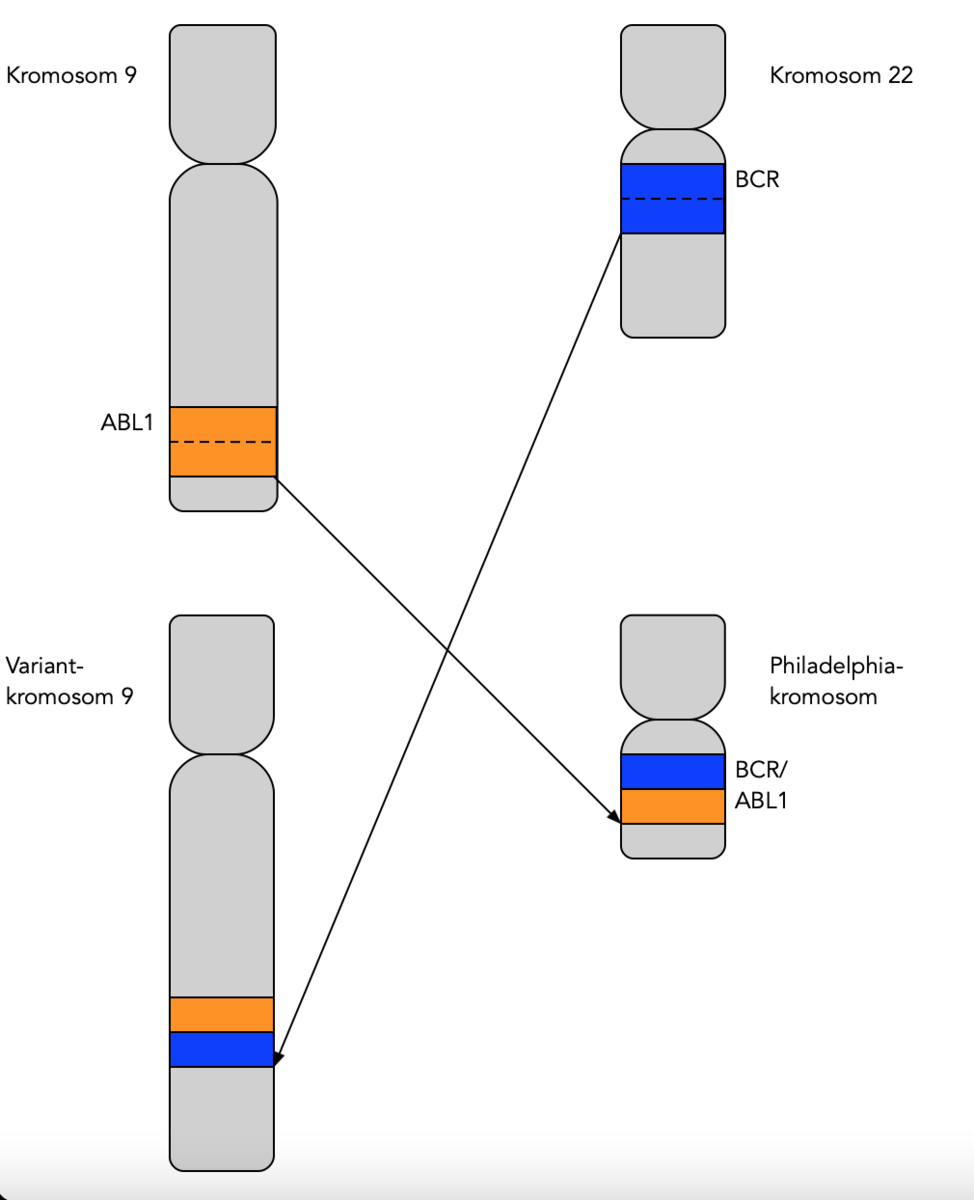
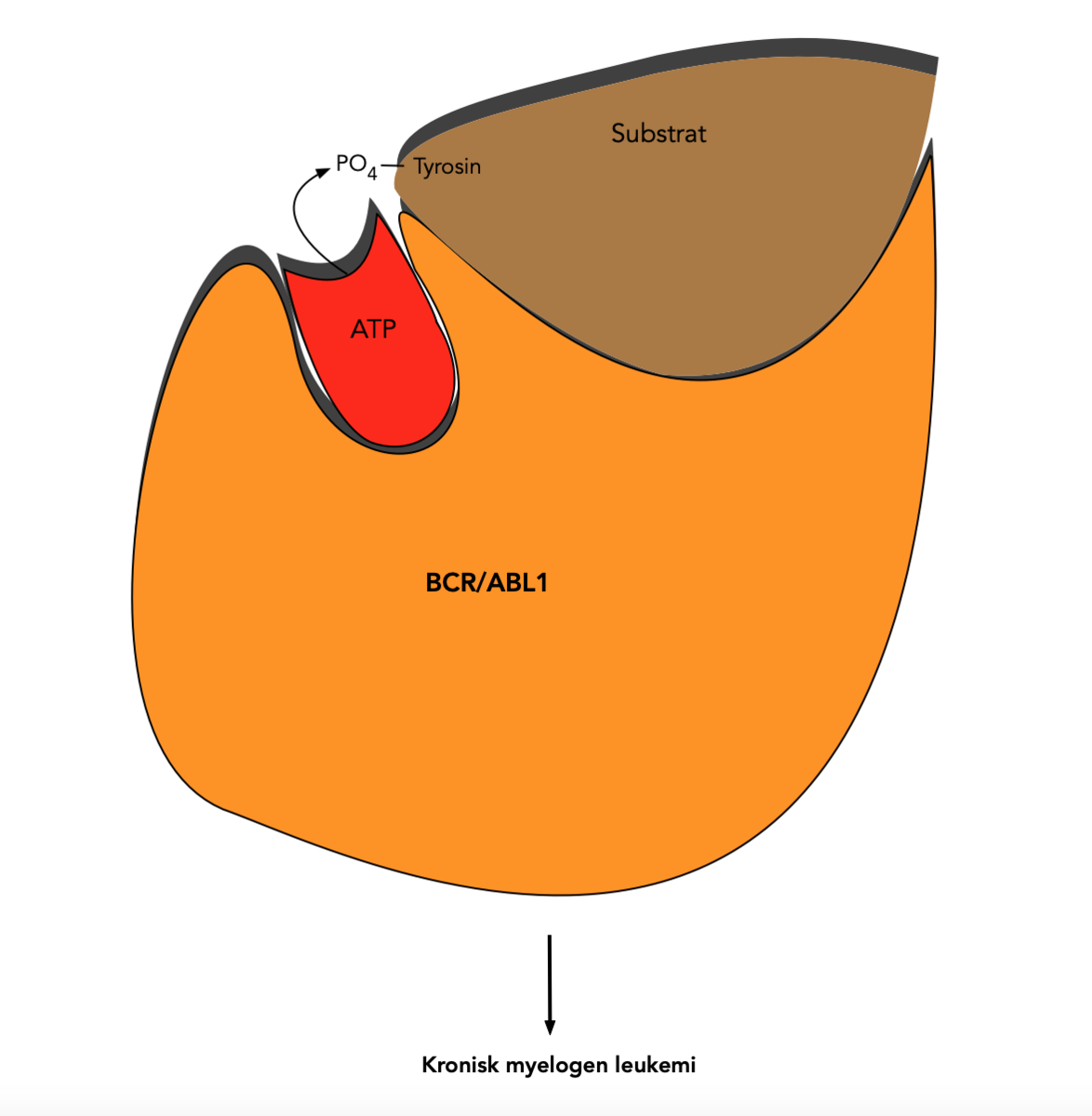
I 1973 ble det påvist at Philadelphia-kromosomet skyldtes en gjensidig bytting av genmateriale mellom kromosom 9 og 22 som på fagspråket kalles translokasjon (9;22). I 1984–1985 ble bruddstedene i DNA-tråden nærmere identifisert, og det ble klart at det ble laget et helt nytt gen på grunn av den nye fusjonen av DNA. Dette fusjonsgenet på kromosom 22 ble kalt for BCR-ABL, hvor BCR står for «breakpoint cluster region» og ABL fra kromosom 9 fikk betegnelsen fra Abelson som var navnet på pasienten hvor dette ble beskrevet. Proteinet som lages fra dette genet viste seg å ha tyrosinkinase-aktivitet, det vil si at det er et enzym som overfører en fosfatgruppe til tyrosin. Fosforylering er en svært vanlig kjemisk reaksjon som aktiverer mange signalveier i cellene. I 1990 ble det gjort eksperimenter hvor det nye fusjonsgenet ble satt inn i mus (transgene mus). Brikkene falt på plass da det viste seg at musene fikk celler i beinmargen som var typiske for KML. Med denne rekken av oppdagelser og eksperimenter ble det slått fast at den nye tyrosinkinasen som ble laget på grunn av translokasjon (9;22) var helt sentral i den molekylære mekanismen som fører til KML.
Det neste naturlige steget var å utvikle en medisin som kunne blokkere effekten av den kreftfremkallende tyrosinkinasen. Etter ti års arbeid rettet mot å blokkere tyrosinkinase-aktiviteten og etterfølgende utprøving på pasienter, ble tyrosinkinasehemmeren imatinib godkjent og markedsført i 2001.
Historien om KML representerer et av de store fremskrittene innen kreftmedisinen. Det er også en fascinerende historie om hvorledes enkle observasjoner, ledsaget av hypoteser og basalforskning med datidens metoder førte frem til en enkel behandling som drastisk endret prognosen av KML 40 år etter de første oppdagelsene. Etter 2001 kom det en oppblomstring av forskning på tyrosinkinasenes rolle ved kreft i håp om å finne andre tilsvarende årsaksmekanismer ved andre kreftsykdommer. Det er funnet en rekke kinaser som har en delvis virkning ved kreft, men de aller fleste krefttypene har sammensatte mekanismer som inntrer etter hverandre og er svært vanskelig å avdekke. KML står i en særstilling når det gjelder enkel årsaksmekanisme. Philadelphia-kromosomet var den første påvisningen av et kromosomavvik som kunne knyttes til kreft. Senere er det funnet tusenvis av kromosomavvik og andre mutasjoner ved kreftsykdommene, men svært få har en så klar årsakssammenheng som kan utnyttes i behandlingen. Selv om den molekylære mekanismen for KML nå er godt kjent, vet vi ikke hvorfor byttingen av DNA-materiale mellom kromosom 9 og 22 skjer. Foreløpig må vi regne med at dette skyldes tilfeldigheter.


Kommentarer
Kommentarer til artikkelen blir synlig for alle. Ikke skriv inn sensitive opplysninger, for eksempel helseopplysninger. Fagansvarlig eller redaktør svarer når de kan. Det kan ta tid før du får svar.
Du må være logget inn for å kommentere.