

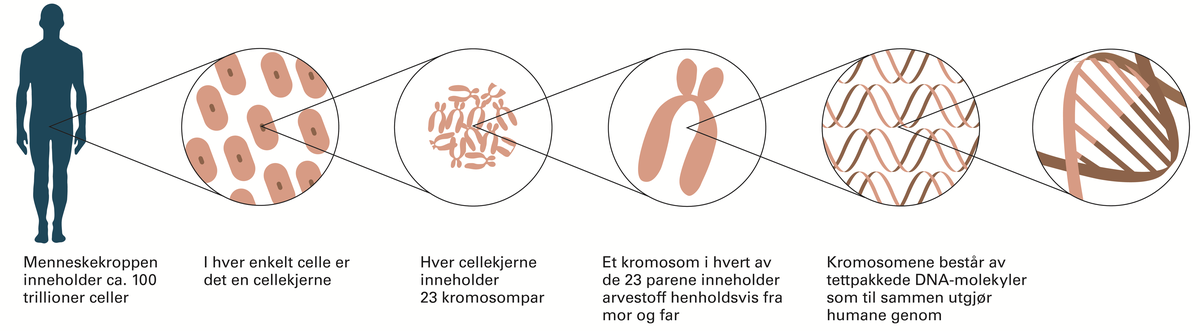
Medisinsk genetikk er læren om arvelige sykdommer og genetiske bidrag til sykdom hos mennesker. Medisinsk genetikk er dermed den delen av humangenetikk som omhandler sykdom og sykdomsmekanismer. Skillet mellom normale genetiske egenskaper og genetisk sykdom kan være flytende. Begge deler skyldes ulike varianter i kromosomenes eller genenes struktur eller funksjon, og genetiske faktorer kan virke alene eller i samspill med andre, ytre faktorer.
Faktaboks
- Også kjent som
- medisinsk arvelære
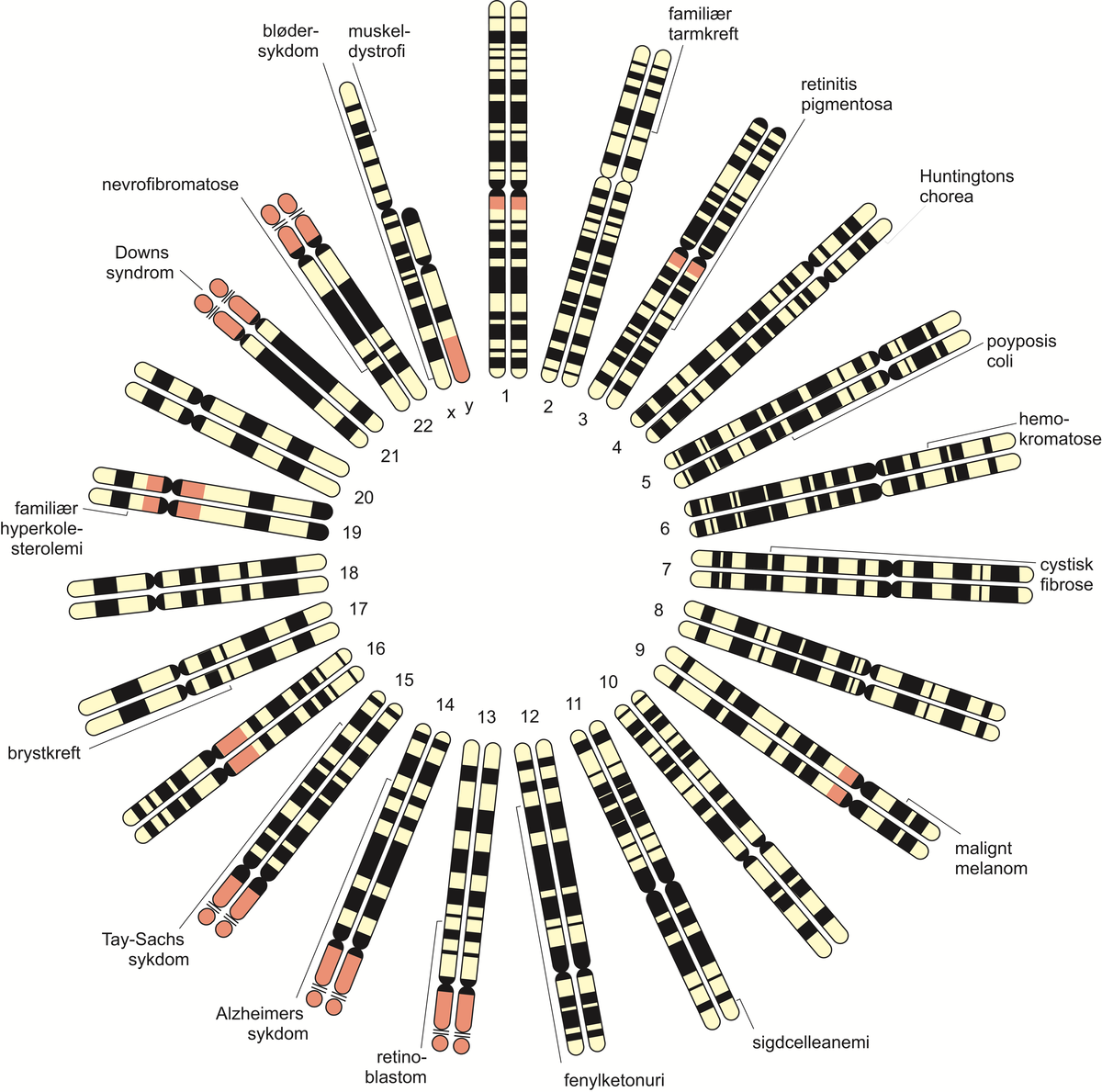
Mennesket har antakelig rundt 25 000 gener. I 2021 var bare i overkant av 4500 gener, altså mindre enn en femtedel, så langt blitt forbundet med en kjent genetisk sykdom eller egenskap. Genenes effekt på sykdom er bare delvis kartlagt.
- Les mer om menneskets genetikk: humangenetikk
Undersøkelse av sykdomsgener er bare én del av faget medisinsk genetikk. Klinisk utredning, tolkning av familiehistorie og genetisk veiledning er også sentralt i utøvelse av faget medisinsk genetikk.
I Norge er medisinsk genetikk en egen spesialitet for leger. Arbeidet til en slik legespesialist omfatter både genetiske laboratorieundersøkelser og klinisk genetikk. Klinisk genetikk er betegnelsen på anvendelse av medisinsk genetikk i direkte møte med pasienter.

Kommentarer
Kommentarer til artikkelen blir synlig for alle. Ikke skriv inn sensitive opplysninger, for eksempel helseopplysninger. Fagansvarlig eller redaktør svarer når de kan. Det kan ta tid før du får svar.
Du må være logget inn for å kommentere.