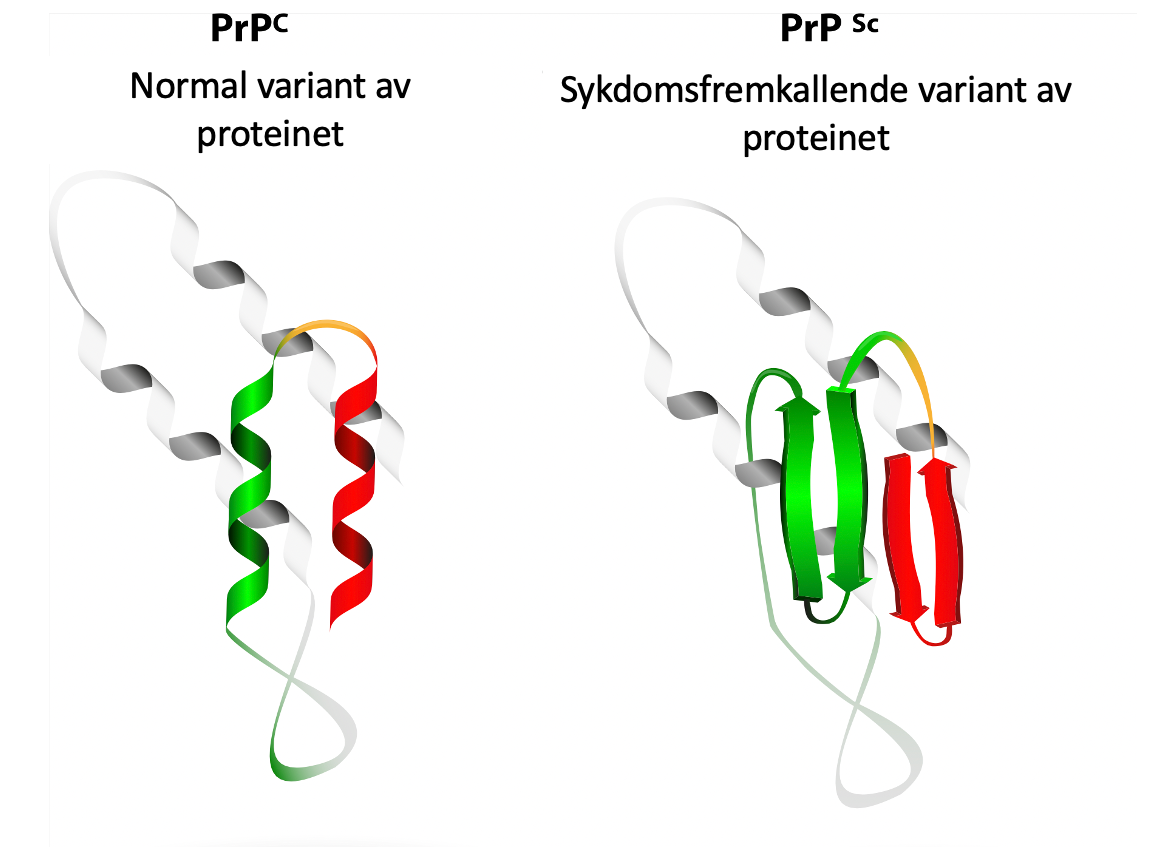
Sykdommen skyldes prioner, som kan påvises i hjernevevet. Prioner er små infeksiøse proteiner, som er en misfoldet variant av prionproteinet (forkortet PrP) og som normalt finnes hos alle mennesker.
Creutzfeldt-Jakobs sykdom (CJS) kan oppstå:
- sporadisk (spontan mutasjon) (sporadisk CJS)
- arvelig (familiær CJS)
- ved smitte fra infiserte dyr eller mennesker (variant CJS)
- ved smitte fra instrumenter i forbindelse med medisinske undersøkelser eller behandlinger (iatrogen CJS)
De langt fleste sykdomstilfellene er sporadiske, det vil si tilfeldig oppståtte mutasjoner i prionproteinet (PrP). Risikoen for dette øker med alderen.
Noen tilfeller er arvelige. Det innebærer at man har arvet en mutasjon i genet som koder for prionproteinet. Dette genet er lokalisert på kromosom 20. Slike arvelige prionsykdommer inkluderer også fatal familiær insomni og Gerstmann-Sträussler-Scheinkers sykdom.
I noen tilfeller kan mennesker bli smittet fra infiserte dyr eller mennesker. Ved smitte fra storfe med kugalskap kan mennesker få en variant av Creutzfeldt-Jakobs sykdom (vCJS). Tidligere ble det sett en variant som kalles kuru-sykdom i Papua Ny-Guinea i forbindelse med rituell kannibalisme.
I noen få tilfeller er sykdommen overført ved medisinske inngrep (såkalt iatrogen smitte), ved hornhinnetransplantasjon, ved implantering av hjerneelektroder tidligere brukt av Creutzfeldt-Jakobs-pasienter, eller tilførsel av humant veksthormon. Det antas at det overførte infeksiøse agenset stammer fra sentralnervesystemet til en syk pasient eller en pasient som er i inkubasjonstiden.

Kommentarer (4)
skrev Jørn Våge
Hei, denne delen av teksten er etter mitt skjønn ikke i henhold til dagens forståelse av prioner:
..., men man tror at sykdommen skyldes en viruslignende organisme som tilhører den gruppen som fremkaller de såkalte «slow virus infections» i hjernen.
svarte Ida Scott
Hei! Du må gjerne legge inn et endringsforslag til artikkelen. Vennlig hilsen Ida Scott, redaksjonen.
svarte Jørn Våge
Hei, mitt forslag er å stryke dette:
, men man tror at sykdommen skyldes en viruslignende organisme som tilhører den gruppen som fremkaller de såkalte «slow virus infections» i hjernen.
Det er henvisning til https://sml.snl.no/prion i teksten foran, og den referansen er tilstrekkelig for korrekt forståelse av agens.
mvh Jørn Våge
svarte Ida Scott
Takk! Gjør det gjerne selv ved å klikke "foreslå endringer i artikkelen". Mvh Ida Scott.
Kommentarer til artikkelen blir synlig for alle. Ikke skriv inn sensitive opplysninger, for eksempel helseopplysninger. Fagansvarlig eller redaktør svarer når de kan. Det kan ta tid før du får svar.
Du må være logget inn for å kommentere.